摘要:繼生產、流通、使用環節的抽檢之后,醫療器械臨床試驗抽檢已經在全國各地開展。以下是2016年至今2018年總局三年內的醫療器械臨床抽檢公告進行了總結。

一、硝煙彌漫的戰場
2016年,CFDA抽取20個項目開展臨床試驗現場檢查,8個存在真實性問題。
2017年,CFDA抽取19個項目開展臨床試驗現場檢查,3個存在真實性問題。
2018年,截止目前,CFDA抽取20個項目開展臨床試驗現場檢查,9個有合規性問題,未公布有真實性問題(請注意第二批10個項目結果尚未公布,但我司項目已傳佳績)。
各省局臨床核查也已開展多年,如廣東、北京、上海、江蘇、廣西、浙江、湖南、天津、遼寧、河南、安徽、四川、山西……
不幸的是:在核查通告中,我們不難發現IVD問題較多,是“重災區”。
我們還發現:目前開展的臨床試驗監督抽查不僅關注的是在注冊項目,還包括備案后現處于試驗中的項目。
由此可見,監管部門對臨床核查有著強大的決心、恒心、專心。
寫到此,心情沉重,小編覺得“質量保證,準時交付”不僅是我們給客戶、公司的承諾,更是我們部門給自己的“軍令狀”:
a)確保臨床進度時,同時也要做好質量控制和質量保證;
b)法規要求絕不是“優秀線”,勉強算上“及格線”;
c)臨床試驗的質量不是操作出來的,而是設計出來的;
d)加強臨床試驗管理,才能保證試驗規范,才能使得產品安全性和有效性可信,才能維護好受試者的權益。
二、臨床試驗質量控制法規依據
《醫療器械監督管理條例》的決定 (國令第680號)
《體外診斷試劑注冊管理辦法》(局令第5號)
《醫療器械注冊管理辦法》(局令第4號)
《醫療器械臨床試驗質量管理規范》(局令第 25號)
《體外診斷試劑臨床實驗指導原則》(2014年16號通告)
《體外診斷試劑臨床試驗指導原則(征求意見稿)》(2018年11月22日)
《醫療器械臨床試驗規定》(局令第5號)(廢止)
《總局關于開展醫療器械臨床試驗監督抽查工作的通告》(2016年第98號)
《國家藥監局綜合司關于印發醫療器械臨床試驗檢查要點及判定原則的通知》(藥監綜械注〔2018〕45號)
三、臨床試驗監督抽查內容
目前臨床監督抽查主要是圍繞臨床試驗的真實性和合規性開展。依據 2018年11月28日發布《醫療器械臨床試驗檢查要點及判定原則》(藥監綜械注〔2018〕45號),根據檢查發現的問題,檢查結果按以下原則判定。
3.1 有以下情形之一的,判定為存在真實性問題:
(1)編造受試者信息、主要試驗過程記錄、研究數據、檢測數據等臨床試驗數據,影響醫療器械安全性、有效性評價結果的;
(2)臨床試驗數據,如入選排除標準、主要療效指標、重要的安全性指標等不能溯源的;
(3)試驗用醫療器械不真實,如以對照用醫療器械替代試驗用醫療器械、以試驗用醫療器械替代對照用醫療器械,以及以其他方式使用虛假試驗用醫療器械的;
(4)瞞報與臨床試驗用醫療器械相關的嚴重不良事件和可能導至嚴重不良事件的醫療器械缺陷、使用方案禁用的合并用藥或醫療器械的;
(5)注冊申請的臨床試驗報告中數據與臨床試驗機構保存的臨床試驗報告中的數據不一致,影響醫療器械安全性、有效性評價結果的;
(6)注冊申請的臨床試驗統計分析報告中數據與臨床試驗統計數據庫中數據或分中心臨床試驗小結中數據不一致,影響醫療器械安全性、有效性評價結果的;
(7)其他故意破壞醫療器械臨床試驗數據真實性的情形。
3.2 未發現真實性問題的,但臨床試驗過程不符合醫療器械臨床試驗相關規定要求的,判定為存在合規性問題。
3.3 未發現上述問題的,判定為符合要求。
四、監督抽查總結
下圖是2016~2018年臨床監督抽查公告中的常見問題分析。其中“核心文件”的整理是最令人頭疼的,此外,“物資管理”、“試驗數據”也是臨床真實性核查中最常見的問題。
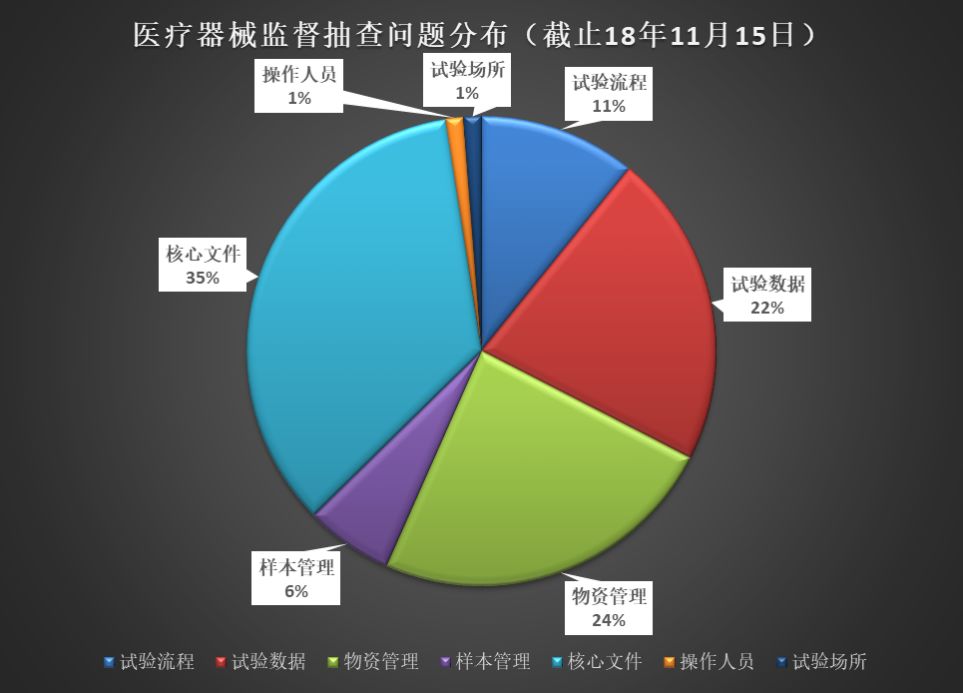
“核心文件”——藥監局是有發目錄清單的(總局關于發布《醫療器械臨床試驗倫理審查申請與審批表范本》等六個文件的通告(2016年第58號):附件6.醫療器械臨床試驗應當保存的基本文件目錄)。文件工作是最基本的工作,實際檢查出來的情況確讓人堪憂。如果連“文件工作”都做不好,藥監局的老師們如何相信臨床試驗的合規性、真實性呢?
基于此,也請大家延伸思考2個問題:
a) 每一個臨床試驗參與者,會不會是另一個臨床試驗的受試者呢?
b) 每一個臨床試驗參與者,會不會又是另一個產品經過臨床試驗并被批準上市的最終使用者呢?
“物資管理”和“試驗數據”——均可以通過文件和記錄體現。《醫療器械臨床試驗質量管理規范(25號令)》要求如下:
第78條所述“臨床試驗結束時,研究者應當確保完成各項記錄、報告。同時,研究者還應當確保收到的試驗用醫療器械與所使用的、廢棄的或者返還的數量相符合,確保剩余的試驗用醫療器械妥善處理并記錄存檔”;
第80條所述“在臨床試驗中,研究者應當確保將任何觀察與發現均正確完整地予以記錄,并認真填寫病例報告表”;
第82條所述“申辦者應當準確、完整地記錄與臨床試驗相關的信息”。
通告中顯示不少項目缺失研究產品“交接記錄”、“運輸記錄”、“儲存條件”、“分發記錄”、“使用記錄”、“回收/銷毀記錄”,甚至還有記錄顯示試驗用樣本“已經超過有效期”,我們不禁要問“真的有做過臨床試驗嗎?”
另外,同一個項目在不同臨床試驗機構被檢查出的問題基本是一致的。究其根本原因,需要考慮下是否是——臨床試驗申辦方及其代表(如CRO)對于臨床試驗態度的問題了——有些文件不是有就行了,如果忽略了時間關系和邏輯順序,那么低級錯誤就會頻繁發生。
隨著監管力度越嚴,如果大家還是抱有僥幸心理并企圖蒙混過關,總有一天會自食惡果,因為“出來混終是要還的”。
小編在工作中無意發現,有些機構老師根據通告做了《申辦方與醫院“黑名單”》,聲稱不與他們合作。碰到這種中心,小編認為還是應該好好珍惜,當以自勉,互相學習,早日進步為宜。
五、通告案例問題歸納
5.1 臨床試驗機構方面
未經審核同意在臨床試驗機構以外場所進行試驗。
5.2 臨床試驗中實施者/申請人的職責履行情況
有非臨床試驗機構人員參與試驗設備操作。
5.3 臨床試驗的批準備案情況
(1) 研究者未全程參與臨床試驗方案的制定過程;
(2) 臨床試驗方案修訂未經醫院倫理委員會、申辦方、主要研究者批準;
(3) 方案無主要研究者、統計學負責人簽字或無機構蓋章;
(4) 方案無臨床試驗主管部門意見、蓋章。
5.4 倫理審查方面
(1) 倫理委員會批準的知情同意書、CRF內容與執行的知情同意書、CRF內容不一致;
(2) 需要快審資料,未提供主審委員的快審審查意見;
(3) 倫理批件未見倫理委員會會議簽到表;
(4) 倫理批件未標明臨床試驗方案和知情同意書的版本號;
(5) 知情同意書中缺少風險告知內容;
(6) 無合理理由家屬代簽知情同意書。
5.5 臨床試驗準備情況
(1) 未按照試驗用醫療器械的預期用途進行方案設計;
(2) 臨床試驗方案編制不嚴謹,前后不一致或者相互矛盾;
(3) 《醫療器械臨床試驗須知》中無受試產品的技術指標等;
(4) 病例未按照統計學原理進行選取,且抽查病例的干擾病例指標無法溯源;
(5) 未根據臨床試驗方案制定標準操作規程;
(6) 未提供臨床試驗質控品交接和檢測記錄;
(7) 未提供對照產品的相關資質文件;
(8) 多中心試驗中,參比試劑不同;
(9) 臨床試驗記錄、表格、文件等資料未簽署具體意見或未蓋章。
5.6 臨床試驗實施情況
(1) 未嚴格按照制定的臨床試驗方案開展試驗;
(2) 受試者入組前未按方案要求完成全部檢測;
(3) 未開展預實驗;
(4) 監查方案可操作性不強,未能在監查中及時發現問題。
(5) 未記錄合并用藥;
(6) 臨床試驗樣本的存儲條件與實際不一致;
(7) 對比試劑的適用機型與試驗用儀器不符。
5.7 臨床試驗數據管理
(1) 原始病歷中相關記錄不全;
(2) 臨床試驗相關圖像評估記錄不全;
(3) 病例臨床試驗原始記錄未記錄修改原因和時間;
(4) 樣本無篩選記錄;
(5) 項目培訓記錄不完全或缺失;
(6) 監查記錄不完全或缺失;
(7) 病例報告表記錄不符合要求;
(8) 病例報告表表中數據與原始記錄不一致;
(9) 臨床試驗檢測數據無操作者、復核者簽字確認等;
(10) 漏報不良事件、嚴重不良事件、器械缺陷等;
(11) 剔除數據無依據和說明;
(12) 未記錄受試產品的失效日期等;
(13) 臨床試驗用設備使用記錄缺失。
5.8受試產品的管理
(1) 通過現場檢查和調取注冊申請資料還發現,該注冊申請項目在開展臨床試驗前未提交該產品的型式試驗報告;
(2) 未提供受試產品、對照產品的分發和回收記錄;
(3) 儀器交接記錄未保留快遞單號;
(4) 試驗用樣本試劑交付給臨床試驗機構時已經超過有效期。
5.9 臨床試驗用樣本的管理
(1) 樣本重復使用,未提供相應說明;
(2) 樣本采集、儲存、分發、使用、留樣、銷毀記錄不完全或缺失;
(3) 樣本類型不符、無法追溯。
5.10 申報資料的情況
(1) 倫理委員會保存的方案與注冊申報材料中的方案病例數和病例分配不一致;
(2) 提交的注冊申請中的臨床試驗方案、報告與臨床試驗機構保存的臨床試驗方案、報告簽章不一致;
(3) 臨床試驗用產品與注冊申報資料的臨床試驗方案和試驗報告中的產品為不一致;
(4) 部分臨床數據與現場提供的統計分析數據不一致等;
(5) 統計分析報告表未對剔除病例做出說明等;
(6) 臨床機構保存的可溯源的隨訪例數與注冊申報資料的臨床試驗總結報告隨訪例數不一致。
小結:限于篇幅問題,小編就先寫到這了。臨床試驗現場核查目前只是剛剛拉開帷幕,作為正在或即將踏上臨床試驗征途的法規工作者,我們要“冬練三九,夏練三伏”,穩扎穩打,放眼全局。在進行臨床試驗時,我們要保證質量,不要一味的追求進度而忽視質量,要遵循產品的自身特點和預期用途,要遵循相關法律法規和相關的SOP要求,否則必將前功盡棄、玩火自焚。
以上內容只是小編的幾點薄見,如若存在錯誤的地方,歡迎批評指正。