CE認證是歐盟的產品安全認證,所有進入歐盟市場的醫療器械都必須進行醫療器械CE認證,醫療器械需要滿足的CE指令有《有源植入性醫療器械指令》(AIMDD, 90/385/EEC)、《醫療器械指令》(MDD,93/42/EEC)和《體外診斷器械指令》(IVDD, 98/79/EC)。
CE認證是歐盟的產品安全認證,所有進入歐盟市場的醫療器械都必須進行醫療器械CE認證,醫療器械需要滿足的CE指令有《有源植入性醫療器械指令》(AIMDD, 90/385/EEC)、《醫療器械指令》(MDD,93/42/EEC)和《體外診斷器械指令》(IVDD, 98/79/EC)。
一、CE認證簡介


CE標記是法律標記,可以按一定比例放大和縮小。也可以看成是兩個相交的圓,兩個字母是等高的,字母“E”中間的一劃要比上下兩筆略短少許。CE標記高度不能低于5mm。
二、醫療器械指令簡介
適用于心臟起搏器、可植入的胰島素泵等,已于1993年1月1日生效,1995年1月1日強制實施。
已于1995年1月1日生效,1998年6月14日強制實施,目前已升版為2007/47/EC。
適用于血細胞計數器、妊娠檢測裝置等,已于1998年12月7日生效,2003年12月7日強制實施。
三、93/42/EEC中的定義、范圍
——疾病的診斷、預防、監視、治療或減輕
——損傷或殘障的診斷、監視、治療、減輕或修補
——解剖學和生理過程的探查、替換或變更
——妊娠的控制
醫療器械不是通過藥理學、免疫學或代謝學作用等方式在人體內或人體上達到其預定的主要作用,但這些方式有助于其他功能的實現。
附件:本身雖然不是器械,但由其制造商專門指定與器械一起使用,使其能夠按照制造商預定的器械用途來使用
制造商:是指在以其名義將器械投放市場前負責器械的設計、制造、包裝和標簽的自然人或法人,無論這些工作是自己完成的,還是由第三方代表他完成的。
預期用途:是指根據制造商在標簽、說明書和或宣傳材料中提供的資料對器械預期的用途。
四、基本框架
MDD93/42/EEC指令共有23條款和12附錄。其重點包括:
條款 1.本指令適用于醫療器械及其附件。
條款 2.成員國必須保證醫療器械只有在保證人身安全和健康時才能被投放市場和投入使用。
條款 3.能保證安全和健康的器械是指滿足附錄I中基本要求的器械。
條款 4.帶有CE標識的醫療器械可以在歐盟市場自由流通。特殊用途的器械不應加貼CE標志:做臨床試驗的器械和定制器械。
條款 5.符合協調標準的醫療器械被認為滿足基本要求。
條款 8.如果發現器械不安全的話,保護條款允許某個成員國采取行動,召回已上市產品,或禁止、限制其投放市場。
條款 9.依據附錄IX進行分類,分為I、IIa、IIb、III類。
條款 10.上市后事故報告的要求,執業醫師和醫療機構,制造商和歐盟代表都應向主管當局報告。
條款 11.醫療器械必須經過某一程序(附錄II—VII)以證明其滿足基本要求。
條款 12.有關系統和打包的醫療器械上市的特殊程序
條款 14.負責將器械投放市場的人員注冊,歐盟數據庫
條款 15.臨床試驗:參照附錄X
條款 17.符合基本要求并通過了相應的符合性評價程序的醫療器械必須帶有CE標志,參照附錄XII
五、附 錄
附錄1.基本要求
附錄2.全面質量保證體系
附錄3.產品型式檢驗
附錄4.產品驗證
附錄5.生產質量保證體系
附錄6.產品質量保證體系
附錄7.自我符合性聲明
附錄8.特殊用途的器械聲明
附錄9.分類規則
附錄10.臨床試驗
附錄11.選擇公告機構準則
附錄12.合格的CE標志
六、產品分類
分類規則的應用由器械的預期用途決定
如果器械是和其他器械配合使用, 分類規則分別適用于每種器械附件
可以和其他一起使用的器械分開獨自分類
啟動或影響某種器械的軟件與器械本身屬于同一類型,獨立的軟件屬于有源醫療器械
如果幾條規則適用于同一器械,以及這些規則的應用會導致不同的分類標準,則規則適用于導致最高分類級別
特別規則(規則13-18)優先于其他規則
時間:暫時(<60分鐘)
短期(<30天)
長期(>30天)
創傷性:非創傷 通過人體孔徑插入
外科創傷 植入
適用位置: 中央循環系統
中樞神經系統
人體孔徑
其他地方
能量供應:無源 有源
規則1---8. 無源醫療器械的分類
規則9---12. 有源醫療器械的分類
規則13---18.特殊醫療器械的分類
規則1~4. 所有非創傷性器械均屬I類,除非它們:
用于存儲體液(血袋例外) | IIa類 |
與IIa或更高類型的有源醫療器械連接使用 | IIa類 |
改變體液成分 | IIa/IIb類 |
一些傷口敷料 | IIa/IIb類 |
規則5. 侵入人體孔徑的醫療器械(不與有源醫療器械相連或僅與I類有源器械相連接)
暫時使用(例如:牙科診斷和治療時使用的手持鏡、牙科加壓材料、胃減壓管、灌腸器械、檢查手套) | I類 |
短期使用(例如:氣管導管、矯正隱形眼鏡、導尿管) | IIa類 |
長期使用(例如:矯正隱形眼鏡、導尿管) | IIb類 |
規則6. 外科創傷性器械暫時使用(如縫合針),屬IIa類,以下情況除外:
可重復使用的外科器械(例如:鉗子、斧子) | I類 |
與中央循環系統或中樞神經系統接觸的器械 | III類 |
可以被吸收的 | IIb類 |
與有源器械連接向人體發送能量或藥物 | IIb類 |
規則7.外科創傷性器械短期使用,屬IIa類,以下情況除外
與中央循環系統或中樞神經系統接觸的器械 | III類 |
產生生物學影響或可以被吸收的 | III類 |
與有源器械連接向人體發送能力或藥物 | IIb類 |
規則8.外科創傷性器械長期使用(接骨板、眼內晶體) IIb類,以下情況除外
安裝在牙齒上的器械 | IIa類 |
與中央循環系統或中樞神經系統接觸的器械 | III類 |
可以被吸收的 | III類 |
向人體給藥物裝置 | III類 |
2003/12/EC乳房植入 | III類 |
2005/50/EC髖關節,膝關節,肩關節 | III類 |
規則9.用于施藥或換能的有源治療器械
給予或交換能量的有源治療器械,(例如:肌肉刺激器、電鉆、皮膚光療機、助聽器、理療超聲設備) | IIa類 |
以一種潛在的危險方式工作的(例如:嬰兒保溫箱、高頻電刀、超聲碎石器、外科激光器、X光機) | IIb類 |
規則10.診斷用的有源器械
提供能量(例如:核磁共振、超聲診斷儀) | IIa類 |
診斷/監視體內放射藥物分布(例如:r照相機、正電子發射成像儀) | IIa類 |
診斷/監視生理功能(例如:心電圖、腦電圖) | IIa類 |
危險情況下監視生理功能(例如:手術中的血氣分析儀) | IIb類 |
發出發射電離輻射(例如:X射線診斷儀) | IIb類 |
規則11.控制藥物或其物質進出人體的有源器械屬于IIa類
如以一種潛在的危險方式工作(例如:麻醉機、透析機、高壓氧艙) | IIb類 |
規則12.所有其他有源醫療器械屬于I類
(例如:觀察燈、牙科椅、輪椅、牙科用治療燈、記錄處理觀察診斷圖像用的有源器械)
規則13.與藥品或血液制品結合的器械
(例如:含殺精子的避孕套、含抗生素的牙髓材料)屬于III類
規則14.避孕用具
規則15.清洗或消毒的器械
醫療器械 | IIa類 |
侵入或創傷醫療器械(例如:內窺鏡消毒) | IIb類 |
接觸鏡(例如:消毒液、護理液) | IIb類 |
規則16. 用于記錄X射線圖像的器械(例如:X光片、熒光盤)屬于IIa類
規則17. 利用動物組織或衍生物的器械(例如:生物心臟瓣膜、腸線、膠原)屬于III類
規則18. 血袋屬于IIb類
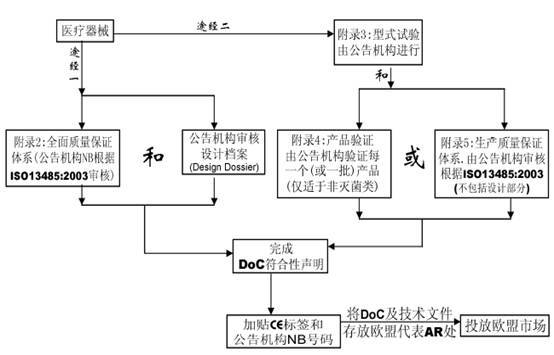
七、合格評定程序
在MDD指令中共有7個合格評定程序,用于該指令條款11中規定的各類器械的合格評定。
附錄2 EC符合性聲明—全面質量保證體系(常用的符合方式)
附錄3 EC產品型式檢驗
附錄4 EC產品驗證
附錄5 EC符合性聲明—生產質量保證體系
附錄6 EC符合性聲明—產品質量保證體系
附錄7 EC符合性聲明—自我符合性聲明
? 附錄8 特殊用途的器械聲明
附錄2 EC符合性聲明—全面質量保證體系(常用的符合方式)
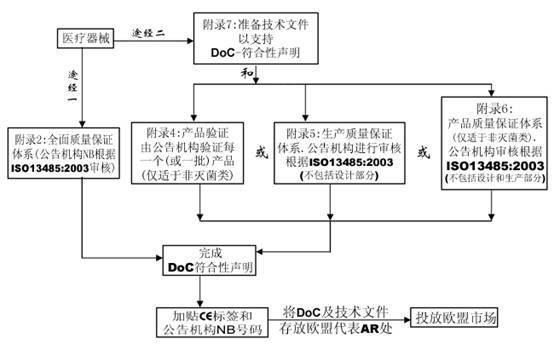
該全面質量保證體系包括產品的設計和生產。它可用于除I類產品外的所有其他產品的合格評定。對于III類產品,設計文檔檢查和該附錄條款4中的認證是必須的,對于IIa類產品,無需設計文檔檢查。
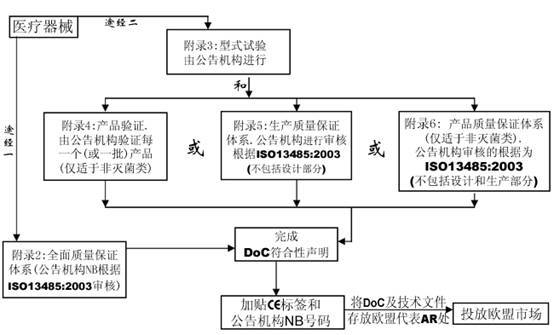
附錄3 EC產品型式檢驗
該附錄描述了產品型式試驗的程序,即制造商向公告機構遞交完整的產品技術文檔以及產品的代表性樣品。公告機構檢查產品是否與技術文檔一致,并是否符合基本要求,如需要則進行這方面的測試,EC型式檢驗證書。該附錄僅包括器械的設計,并適用于IIb或III類醫療器械。
附錄4 EC產品驗證
該EC產品驗證程序確保器械依據一個認可的型號或技術文件中描述的器械生產。在該程序下,公告機構檢查每個或多個樣品并進行試驗以證明產品是否符合已認可文件化的設計。該程序不適用于無菌醫療器械。
附錄5 EC合格聲明—生產質量保證體系
該附錄描述了一個生產質量保證體系,即由公告機構證明該體系能保證器械能夠依據認可的型號,或依據技術文件中描述的器械生產。該附錄適用于IIa, IIb和III類器械。
附錄6 EC合格聲明—產品質量保證體系
該附錄描述了一個質量體系,該體系通過產品的最終檢驗和試驗以確保生產的器械符合已認可的型號,或技術文件中規定的器械。該附錄適用于IIa和IIb類器械。該程序不適用于無菌類醫療器械。
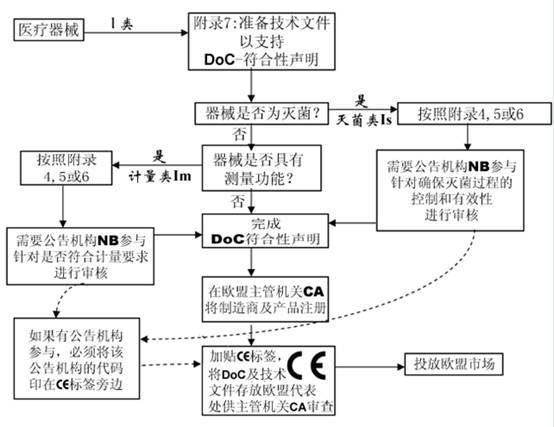
附錄7 EC合格聲明—自我符合性聲明
該附錄描述制造商必須準備技術文件以支持某類醫療器械的合格聲明,這里無需公告機構介入。該附錄適用于I和IIa類器械。
附錄8 特殊用途的器械聲明
該附錄描述對于定制器械和臨床調查類器械,制造商必須準備技術文件以支持醫療器械的合格聲明,這里無需公告機構介入。
八、CE認證的基本流程
1.分析該器械的特點,確定它是否在指令范圍內
2.確定該器械的分類類別
3.確認適用的基本要求/有關的協調標準
4.確認該器械滿足基本要求/協調標準,并使證據文件化(技術文檔的整理)
5.確定相應的符合性評價程序
6.對于IIa類或更高類型器械,以及I類無菌或測量器械,應通過公告機構并進行符合性評價程序
7.起草符合性聲明并加貼CE標志
I 類
IIa 類
IIb 類
III 類